This information sheet has been developed by the Mucopolysaccharide & Related Diseases Society of Australia (the MPS Society) to provide information about mucopolysaccharidosis type II (MPS II), its clinical presentation and medical management.
The content of the information sheet draws on the experiences of parents and doctors with reference to the medical literature. It is not intended to replace medical advice or care.
For reference purposes, it may be useful to provide a copy of this information sheet to your GP and others who are involved in providing medical or supportive care.
MPS II is an inherited disorder that encompasses a wide spectrum of severity. In some people the brain may be affected in combination with physical symptoms; others may develop physical symptoms with no brain involvement. Physical symptoms may include hearing problems, bone and joint malformation, and heart and breathing difficulties. The rate of disease progression and age of onset vary considerably, even in affected siblings: in some it may be rapid, with diagnosis in the first few years of life; in others it may be relatively slow and diagnosis may not occur until the third or fourth decade. Generally, if clinical symptoms are apparent early in life it is more likely that disease progress will be rapid and the brain is more likely to be affected.
MPS II is also known as Hunter syndrome, so named after the doctor who first described the condition.
In common with the other MPS disorders, the characteristic of MPS II is the build up (or ‘storage’) of long chains of sugar molecules called mucopolysaccharides in the body’s cells. ‘Muco’ refers to the thick jelly-like consistency of the molecules, ‘poly’ means many, and ‘saccharide’ is a general term for the sugar part of the molecule. Mucopolysaccharides are also referred to as glycosaminoglycans (or GAGs for short) but for the purpose of this information sheet, the term mucopolysaccharide will be used.
Mucopolysaccharides are used by the cells to build connective tissues in the body, such as skin, muscle, cartilage and bone. They also help with many other cellular functions, including growth control, organ development and signalling between cells.
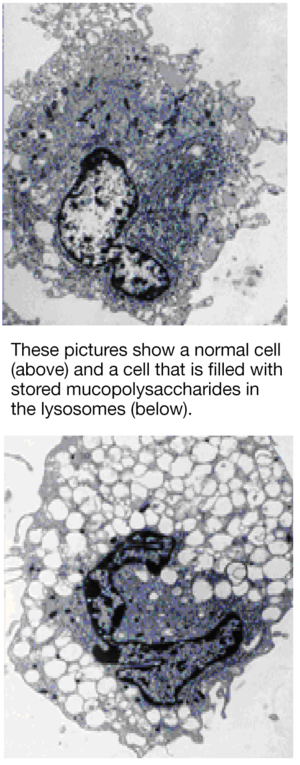
The human body is made up of billions of cells. Each cell contains various structures that carry out many functions important to life. One such structure is known as the lysosome [pronounced lie-so-soam]. Mucopolysaccharides carry out their tasks outside the cell. Once their job is complete they are transported to the lysosomes to be broken down (or degraded) into their basic building blocks. Degradation requires the action of enzymes that are found inside the lysosomes. Once the mucopolysaccharides have been broken down by these enzymes, they are transported out of the lysosomes to be reassembled and re-used to build tissue, etc. Mucopolysaccharides are therefore in a continuous process of being recycled.
In people with an MPS disorder one of the lysosomal enzymes that is needed to degrade mucopolysaccharides is either missing or is present at levels that do not allow the recycling process to work properly. This means that the mucopolysaccharides cannot be completely degraded and removed from the lysosome in the usual way. As a result, partially broken down mucopolysaccharides remain ‘stored’ in the lysosomes: with time, lysosomes increase in size as the amount of storage increases. This interferes with normal cell functioning and causes progressive clinical problems in affected people.
People with MPS II are deficient in a lysosomal enzyme called iduronate-2-sulphatase (or 2-sulphatase or IDS, for short], which is essential in breaking down two mucopolysaccharides called dermatan sulphate and heparan sulphate. The amount of dermatan sulphate storage influences the extent of physical symptoms; the amount of heparan sulphate storage influences whether or not the brain will be affected.
It has been estimated that about 1 in 136,000 male births are affected by MPS II.
The incidence of all MPS disorders combined (of which 11 are currently recognised) is estimated to be 1 in 25,000 births.
The MPS group of disorders belong to a larger group of about 50 inherited disorders collectively known as lysosomal storage disorders, so named because storage of materials that are unable to be properly degraded (mucopolysaccharides in the case of the MPS disorders) occurs in the lysosome. It is estimated that lysosomal storage disorders occur in about 1 in every 5,000 to 7,000 births.
MPS II is inherited in what is called an X-linked recessive manner. The ‘X’ and ‘Y’ chromosomes determine the sex of a person: males have one X chromosome and one Y chromosome (XY), whilst females have two X chromosomes (XX). In MPS II the gene that codes for deficient production of 2-sulphatase enzyme is located on the ‘X’ chromosome. Thus, males who inherit the faulty X chromosome from their mother will be affected; females who inherit the faulty X chromosome will be carriers but generally do not develop symptoms as they have a second, unaffected X chromosome that compensates.
If a woman is a carrier for MPS II, in each pregnancy there is a:
50% (1 in 2) risk that any male born to her will be affected; and a
50% (1 in 2) risk that any female born to her will be a carrier (but generally not affected);
The sisters and maternal aunts of an affected male have a 50% (1 in 2) chance of being carriers and a 25% (1 in 4) overall risk of passing the condition to any male born to them.
If a male with MPS II has children, all of his daughters will be carriers and all of his sons will be unaffected.
Although not common, in some cases MPS II might arise in a generation without the mother being a carrier or there being any family history of the condition.
The MPS Society has produced a specialist booklet (The Pattern of Inheritance) that is available.
Because MPS II is inherited it is important to seek genetic counselling as there may be implications for other children in the family, future pregnancies and extended family members. Geneticists and/or genetic counsellors will explain the inheritance pattern and help determine who should be tested.
At the present time, there is no routine screening procedure to diagnose a newborn baby with MPS II. If there is a family history of the disorder, however, prenatal testing can be arranged in the early stages of pregnancy (see below) or soon after birth. MPS II is not well known in the community. As the initial symptoms are variable it is often not easily recognised by doctors, hence (in the absence of a family history) diagnosis is often made after obvious problems have developed.
To diagnose MPS II, mucopolysaccharides are usually first measured in urine, followed by measurement of enzyme activity in blood. Increased heparan sulphate and dermatan sulphate in urine, and a decrease in the activity of 2-sulphatase enzyme in blood, is usually consistent with a diagnosis of MPS II. To confirm the urine and blood results, it is useful to measure enzyme activity in a small piece of skin.
Diagnosis by mutation testing may also be possible. Mutations are mistakes in the genetic information (DNA) that is inherited by an affected child from their parents. In MPS II, the mutations are present in the gene that codes for 2-sulphatase and lead to a defect in its production. If the disease-causing mutations are found (which is not always possible to achieve) testing future pregnancies or other family members may be simplified. Mutation testing can be done using either blood or skin.
It is generally agreed that a comprehensive medical and supportive care plan should be started as early as possible after diagnosis to promote the best quality of life.
Testing a fetus for an inherited condition whilst it is still in the womb is called prenatal testing and can be performed if there is a family history of the condition.
Prenatal testing is usually done within the first three months of pregnancy. If you are a carrier for MPS II and wish to consider prenatal testing, it is important to discuss it with your doctor, a geneticist or genetic counsellor prior to or during the very early stages of pregnancy.
In common with other MPS disorders, MPS II is progressive, meaning that the symptoms worsen with time.
The biological processes that determine the age at which symptoms appear and the rate at which they progress are complex and not all are clearly understood. Mucopolysaccharide storage begins as a result of mistakes (mutations) in the genetic information (DNA) that code for the production of a specific enzyme that is responsible for breaking down specific mucopolysaccharides. These mutations determine how much active enzyme can be made, which will affect how much mucopolysaccharide can be broken down in the lysosome. As a general rule, if a mutation allows more active enzyme to be made, the mucopolysaccharide can be broken down more efficiently so disease progress is likely to be slower, with less storage occurring;
if a mutation allows little or no active enzyme to be made, mucopolysaccharide break down will be much less efficient and more will remain stored, so disease progress is likely to be more rapid.
Whilst mucopolysaccharide storage is a significant cause of symptoms it is important to understand that it is one part of a complex ‘cascade’ of changes that occur as a result of the reduction in enzyme activity: the mucopolysaccharides cannot be properly broken down in the lysosomes at the correct time and recycled; in turn, this causes abnormal changes to their function as well as to other functions of the cell. The flow-on effects of these changes significantly contribute to clinical outcome and disease progression in addition to the storage itself. Research is continuing to understand this ‘cascade’ of changes to improve diagnosis, predicting the rate of disease progression (prognosis) and treatment options.
It is difficult to be precise about life expectancy because of variation in severity and age of onset. Some individuals whose brain is affected have lived into adulthood but this is usually accompanied by a decline in their quality of life as brain function deteriorates.
If the brain is not affected, a more normal life span can be expected, but significant physical problems can develop that, without treatment, may reduce life expectancy.
MPS II does not affect fertility. Teenagers will go through puberty, although it may be delayed.